On Kolmogorov complexity and why aging is hard
by Kejun Ying
15 March 2026
5 min read
Jonathan Gorard recently wrote a thread arguing that LLMs exposed how much of human knowledge lives at surprisingly low Kolmogorov complexity. The minimal algorithmic representation of most text and code is far more compact than the artifact itself. LLMs succeed because they learn to decompress efficiently.
I think this framing applies to biology, with an important caveat.
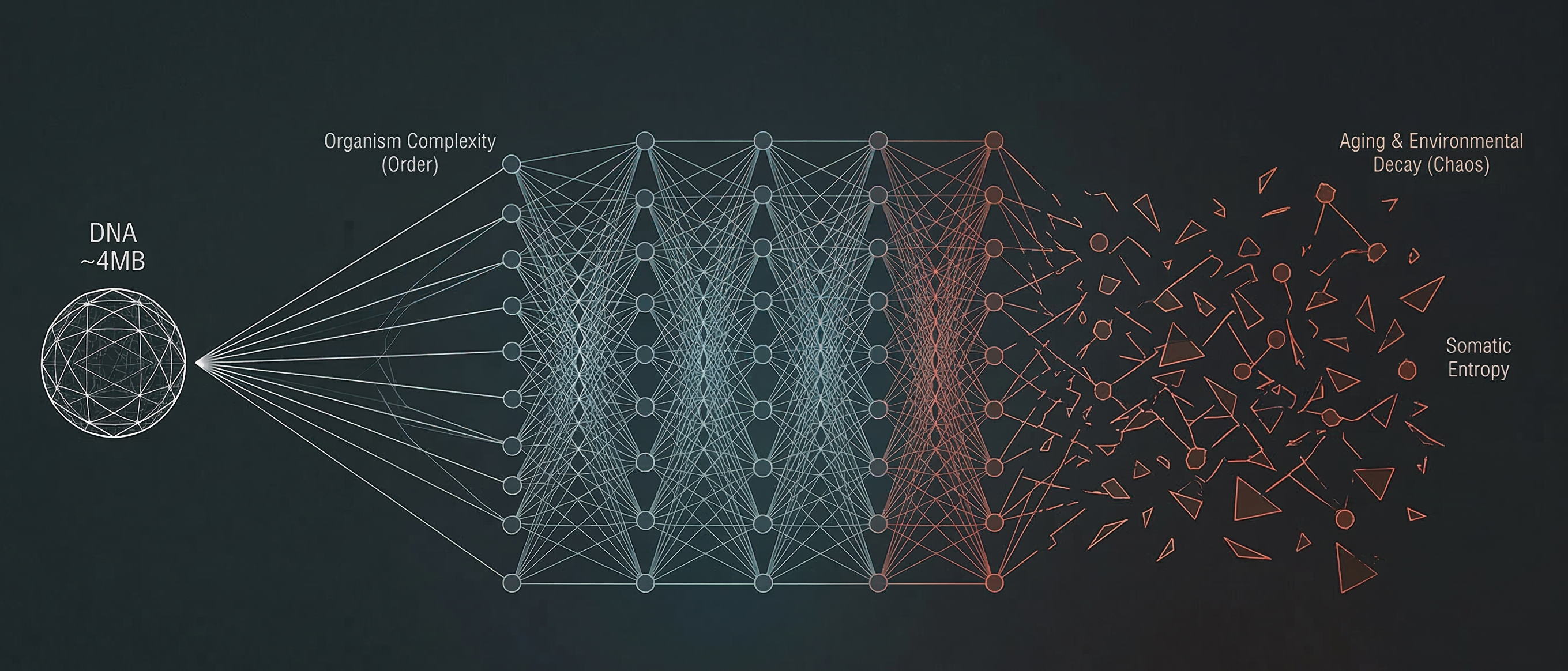
The organism itself is low complexity. The human genome is roughly 750MB of raw sequence, with over 50% consisting of repeats (Lander et al., 2001). Even at 750MB, this is a strikingly compact program for what it builds: 37 trillion cells, hundreds of cell types, decades of coordinated development. Gene regulation, transcription, folding, morphogenesis: all steps in a decompression pipeline that unpacks a compact code into an organism. AlphaFold proved this for proteins (Jumper et al., 2021). Sequence in, structure out. It works because protein structure has low Kolmogorov complexity relative to its apparent complexity. The information is already in the sequence. The model just learns to decompress it.
Where the analogy breaks is aging.
A compact system running inside a high-complexity environment for decades accumulates damage that has no compact description. Every stochastic mutation, every environmental exposure, every protein misfolding event, every immune encounter adds irreducible complexity. Gladyshev formalized this as the “deleteriome”: the cumulative burden of deleterious changes across all levels of biological organization, from molecules to organs, driven by genetic, environmental, and stochastic processes (Gladyshev, 2016). The damage is generated by the interaction between a compact program and an incompressible world. As he once put it: aging might be more complex than life itself. For life, you need to know how to build. For aging, there’s a combinatorial space of things that can go wrong.
Lipsitz and Goldberger showed decades ago that aging is associated with a progressive loss of physiological complexity, from heartbeat dynamics to gait patterns (Lipsitz & Goldberger, 1992). The system becomes less able to respond to perturbation. This loss of complexity at the physiological level is itself the signature of high-complexity damage at the molecular level: the accumulated noise in damaged macromolecules, epigenetic drift, and cellular heterogeneity that Gladyshev’s group has documented (Ogrodnik et al., 2018).
This explains a pattern in medicine. For diseases with a compact cause, you can find a compact fix. Imatinib targets one fusion gene in CML. Yamanaka factors reset cell identity with four transcription factors. These work because the cause is low-complexity, so the cure can be too. Simple causes admit simple cures.
Aging has no simple cure. The corruption is distributed, emergent, high-dimensional.
Consider the two most popular approaches. The pharmaceutical approach targets key regulatory nodes: rapamycin for mTOR, metformin for AMPK, senolytics for senescent cell clearance. These are compact interventions. They can work at specific layers and may extend lifespan by around 10%. But the damage they leave untouched is combinatorially larger than the damage they address. They will not solve aging.
The reprogramming approach goes further. Yamanaka factors can reset epigenetic age in cells (Takahashi & Yamanaka, 2006), and partial reprogramming has shown effects in vivo, including restoring vision in aged mice (Lu et al., 2020). But reprogramming resets epigenetic marks, and epigenetic drift is one layer of aging among many. Misfolded proteins, somatic mutations, lipid peroxidation, and other forms of molecular damage accumulate in parallel. They don’t share the same compact structure that reprogramming can reset. The damage reprogramming cannot reach is the damage that has no compact description. It will not solve aging either.
Compact interventions can only solve the compact subset of aging. The rest of the problem is high-dimensional, distributed, and stochastic. There is no “undo button” that reverses all of it, because the system itself doesn’t know what all the damage is.
If aging has high Kolmogorov complexity, then solving it requires something evolution never built: new biological machinery. Designed proteins, new feedback loops, new pathways that can handle the high-complexity damage that compact interventions cannot reach. This is why I think protein design is where the field ultimately needs to go. You can’t compress an incompressible problem into a compact solution. You need to write new programs.
The foundation model question then becomes something different from what most people in the field are pursuing. The goal should not be to compress a disease phenotype down to a few drug targets. The goal should be to map the full complexity of how biology decays, so we know what new programs to write.
Inspired by Jonathan Gorard’s thread on Kolmogorov complexity and LLMs. The quote from Vadim Gladyshev is from a conversation during my time in his lab at Harvard.
Correction (2026-03-16): An earlier version of this post stated the genome compresses to ~4MB. This was incorrect. The ~4MB figure corresponds to the cross-individual genetic variation (~3 million SNPs at 2 bits each), not the genome itself. The haploid human genome is ~750MB raw (~3.2 billion base pairs at 2 bits each), which can be further compressed due to over 50% repetitive sequence, but remains in the hundreds of megabytes range. The core argument is unchanged: 750MB is still remarkably compact for encoding 37 trillion cells.
References
Lander ES, Linton LM, Birren B, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860-921. doi:10.1038/35057062
Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583-589. doi:10.1038/s41586-021-03819-2
Gladyshev VN. Aging: progressive decline in fitness due to the rising deleteriome adjusted by genetic, environmental, and stochastic processes. Aging Cell. 2016;15(4):594-602. doi:10.1111/acel.12480
Lipsitz LA, Goldberger AL. Loss of ‘complexity’ and aging: potential applications of fractals and chaos theory to senescence. JAMA. 1992;267(13):1806-1809. doi:10.1001/jama.1992.03480130122036
Ogrodnik M, Salmonowicz H, Gladyshev VN. Integrating cellular senescence with the concept of damage accumulation in aging: relevance for clearance of senescent cells. Aging Cell. 2019;18(1):e12841. doi:10.1111/acel.12841
Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126(4):663-676. doi:10.1016/j.cell.2006.07.024
Lu Y, Brommer B, Tian X, et al. Reprogramming to recover youthful epigenetic information and restore vision. Nature. 2020;588:124-129. doi:10.1038/s41586-020-2975-4